Telefone 212 693 530* *Custo de Chamada para a Rede Fixa de acordo com o seu tarifário




Teste de portadores
-
O que é o teste de portadores?
Qualquer pessoa pode ser portadora de variantes (mutações) genéticas sem que o saiba. Os portadores são geralmente pessoas saudáveis, no entanto, correm risco de transmitir uma doença genética aos seus filhos dependendo das mutações das quais são portadores. O teste de portadores é um tipo de teste genético que permite determinar se há risco de transmissão de determinadas doenças genéticas ao seu filho. Este teste pode ser solicitado preferencialmente antes da gravidez e sempre após uma consulta de genética médica.
É o teste indicado para mim?
Grande parte das pessoas, independentemente da sua etnia ou da sua história familiar, não sabe que pode ser portadora de uma variante (mutação) genética até ter um filho afetado com a doença.
- Casais que planeiem uma gravidez e que desejem conhecer o risco de transmissão de mutações genéticas que causem doenças genéticas à descendência;
- Dadores de óvulos e espermatozóides;
- Receptoras de óvulos ou espermatozóides;
- Casais consanguíneos;


O teste de portadores
é o teste de compatibilidade genética mais completo, que combina a sequenciação NGS com outras técnicas de biologia molecular para estudar cerca de 300 doenças genéticas.
- Existência de tratamentos ou possível intervenção precoce;
- Relevância clínica;
- Gravidade;
- Aparecimento precoce da doença;
- Incidência;
Lista de Genes e Doenças
| Gene | Herança | Doença | Origem Populacional | Frequência de Portadores | Taxa de Deteção | Risco Residual Pós-Teste* | Risco pós-Teste da criança afetada** |
|---|---|---|---|---|---|---|---|
| Gene | Herança | Doença | Origem Populacional | Frequência de Portadores | Taxa de Deteção | Risco Residual Pós-Teste* | Risco pós-Teste da criança afetada** |
| ABCA12 | AR | Ictiose Congénita: Relacionada com ABCA12 | Geral | <1/500 | 98% | 1/24951 | <1/49902000 |
| ABCA4 | AR | Doença de Stargardt | Geral | 1/51 | 98% | 1/2501 | 1/510204 |
| ABCB11 | AR | Colestase Intra-hepática Progressiva Familiar: Tipo 2 | Geral | 1/112 | 98% | 1/5551 | 1/2486848 |
| ABCC8 | AR | Hiperinsulinismo Familiar Tipo 1: Relacionado com ABCC8 | Geral Judeu Ashkenazi Finlandês Médio Oriente |
1/112 1/44 1/25 1/25 |
98% 98% 98% 98% |
1/5551 1/2151 1/1201 1/1201 |
1/2486848 1/378576 1/120100 1/120100 |
| ABCD1 | XL | Adrenoleucodistrofia: Ligado ao X | Geral | 1/21000 | 99% | 1/2099901 | 1/8399604 |
| ACADM | AR | Deficiência de Acil-CoA Desidrogenase de Cadeia Média (MCAD) | Geral Este da Ásia Europeu Nativo Americano |
1/69 1/198 1/52 1/43 |
98% 99% 99% 96% |
1/3401 1/19701 1/5101 1/1051 |
1/938676 1/15603192 1/1061008 1/180772 |
| ACADS | AR | Deficiência de Acil-CoA Desidrogenase de Cadeia Curta (SCAD) | Geral Africano Afro-americano Médio Oriente Europeu Sul da Ásia / Índia |
1/85 1/52 1/52 1/52 1/76 1/51 |
99% 99% 99% 99% 99% 99% |
1/8401 1/5101 1/5101 1/5101 1/7501 1/5001 |
1/2856340 1/1061008 1/1061008 1/1061008 1/2280304 1/1020204 |
| ACADVL | AR | Deficiência de Acil-CoA Desidrogenase de Cadeia Muito Longa (VLCAD) | Geral Médio Oriente Nativo Americano Sul da Ásia / Índia |
1/118 1/74 1/61 1/73 |
93% 93% 93% 93% |
1/1672 1/1044 1/858 1/1030 |
1/789184 1/309024 1/209352 1/300760 |
| ACAT1 | AR | Deficiência de 3-Cetotiolase | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| ACOX1 | AR | Deficiência de Acil-CoA Oxidase Peroxissomal | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| ADA | AR | Deficiência Adenosina Desaminase | Geral | 1/224 | 93% | 1/3187 | 1/2855552 |
| ADAMTS2 | AR | Síndrome de Ehlers-Danlos: Dermatosparaxis Tipo VIIC | Geral Judeu Ashkenazi |
<1/500 1/248 |
98% 98% |
<1/24951 1/12351 |
<1/49902000 1/12252192 |
| AGA | AR | Aspartilglucosaminúria | Geral Finlandês |
<1/500 1/71 |
98% 98% |
<1/24951 1/3501 |
<1/49902000 1/994284 |
| AGL | AR | Doença de Armazenamento de Glicogénio: Tipo III | Geral Feroês Inuíte Judeu Norte-africano |
1/158 1/28 1/25 1/37 |
95% 95% 95% 95% |
1/3141 1/541 1/481 1/721 |
1/1985112 1/60592 1/48100 1/106708 |
| AGXT | AR | Hiperoxalúria Primária: Tipo 1 | Geral Europeu |
1/120 1/173 |
99% 99% |
1/11901 1/17201 |
1/5712480 1/11903092 |
| AIRE | AR | Síndrome de Poliendocrinopatia Autoimune tipo I | Geral Finlandês |
1/150 1/79 |
98% 98% |
1/7451 1/3901 |
1/4470600 1/1232716 |
| ALDH3A2 | AR | Síndrome de Sjogren-Larsson | Geral | 1/250 | 98% | 1/12451 | 1/12451000 |
| ALDOB | AR | Intolerância Hereditária à Frutose | Geral Afro-americano Africano Europeu Médio Oriente |
1/122 1/250 1/250 1/67 1/97 |
99% 99% 99% 99% 99% |
1/12101 1/24901 1/24901 1/6601 1/9601 |
1/5905288 1/24901000 1/24901000 1/1769068 1/3725188 |
| ALG6 | AR | Defeitos Congénitos da Glicosilação: Tipo IC | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| ALPL | AR | Hipofosfatasia | Geral Europeu Menonita |
1/158 1/274 1/25 |
95% 95% 95% |
1/3141 1/5461 1/481 |
1/1985112 1/5985256 1/48100 |
| AMH | AR | Síndrome do Ducto Mülleriano Persistente Tipo I | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| AMHR2 | AR | Síndrome do Ducto Mülleriano Persistente Tipo II | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| AMT | AR | Encefalopatia por Glicina: Relacionada com AMT | Geral Finlandês |
1/373 1/117 |
98% 98% |
1/18601 1/5801 |
1/27752692 1/2714868 |
| AR | XL | Síndrome de Insensibilidade Androgénica: Completa | Geral | 1/14286 | 98% | 1/714251 | 1/2857004 |
| ARG1 | AR | Deficiência de Arginase | Geral | 1/296 | 98% | 1/14751 | 1/17465184 |
| ARSA | AR | Leucodistrofia Metacromática | Geral Europeu |
1/100 1/78 |
95% 95% |
1/1981 1/1541 |
1/792400 1/480792 |
| ARSB | AR | Mucopolissacaridose Tipo VI (Síndrome de Maroteaux-Lamy) | Geral Australiano Ocidental |
1/250 1/283 |
98% 98% |
1/12451 1/14101 |
1/12451000 1/15962332 |
| ASL | AR | Deficiência de Argininosuccinato Liase | Geral | 1/132 | 90% | 1/1311 | 1/692208 |
| ASNS | AR | Deficiência de Asparagina Sintetase | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| ASPA | AR | Doença de Canavan | Geral Judeu Ashkenazi |
1/300 1/55 |
97% 96% |
1/9968 1/1351 |
1/11961600 1/297220 |
| ASS1 | AR | Citrulinémia: Tipo I | Geral Este da Ásia |
1/119 1/132 |
96% 96% |
1/2951 1/3276 |
1/1404676 1/1729728 |
| ATM | AR | Ataxia-Telangiectasia | Geral | 1/100 | 92% | 1/1239 | 1/495600 |
| ATP6V1B1 | AR | Acidose Tubular Renal com Surdez | Geral | 1/500 | 98% | <1/24951 | <1/49902000 |
| ATP7A | XL | Doença de Menkes | Geral | 1/50000 | 99% | 1/4999901 | 1/19999604 |
| ATP7B | AR | Doença de Wilson | Geral Europeu Judeu Ashkenazi |
1/87 1/42 1/70 |
98% 98% 98% |
1/4301 1/2051 1/3451 |
1/1496748 1/344568 1/966280 |
| BBS1 | AR, DG | Síndrome de Bardet-Biedl: Tipo 1 | Geral | 1/367 | 99% | 1/36601 | 1/53730268 |
| BBS10 | AR, DG | Síndrome de Bardet-Biedl: Tipo 10 | Geral | 1/395 | 99% | 1/39401 | 1/62253580 |
| BBS12 | AR, DG | Síndrome de Bardet-Biedl: Tipo 12 | Geral | 1/791 | 99% | 1/79001 | 1/249959164 |
| BBS2 | AR, DG | Síndrome de Bardet-Biedl: Tipo 2 (+) | Geral Judeu Ashkenazi |
1/621 1/107 |
99% 99% |
1/62001 1/10601 |
1/154010484 1/4537228 |
| BBS2 | AR, DG | Retinite Pigmentosa 74 (+) | Geral Judeu Ashkenazi |
1/621 1/107 |
99% 99% |
1/62001 1/10601 |
<1/10000000 1/4537228 |
| BBS2 | AR, DG | Doenças Relacionadas com BBS2 | Geral Judeu Ashkenazi |
1/621 1/107 |
99% 99% |
1/62001 1/10601 |
1/154010484 1/4537228 |
| BCHE | AR | Deficiência de Butirilcolinesterase | Geral | 1/28 | 99% | 1/2701 | 1/302512 |
| BCKDHA | AR | Doença da Urina de Xarope de Bordo Tipo IA | Geral Menonita |
1/321 1/10 |
98% 98% |
1/16001 1/451 |
1/20545284 1/18040 |
| BCKDHB | AR | Doença da Urina de Xarope de Bordo Tipo IB | Geral Judeu Ashkenazi |
1/364 1/97 |
98% 98% |
1/18151 1/4801 |
1/26427856 1/1862788 |
| BCS1L | AR | Síndrome de Björnstad (+) | Geral | <1/500 | 98% | 1/24951 | <1/49902000 |
| BCS1L | AR | Síndrome de GRACILE (+) | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| BCS1L | AR | Deficiência do Complexo III Mitocondrial (+) | Geral | <1/500 | 98% | 1/24951 | <1/49902000 |
| BCS1L | AR | Condições Relacionadas com BCS1L | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| BLM | AR | Síndrome de Bloom | Geral Judeu Ashkenazi |
1/800 1/134 |
87% 99% |
1/6147 1/13301 |
1/19670400 1/7129336 |
| BRIP1 | AR | Anemia de Fanconi Grupo J | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| BSND | AR | Síndrome Bartter | Geral | <1/500 | 98% | 1/24951 | 1/49902000 |
| BTD | AR | Deficiência de Biotinidase | Geral Europeu Latino Médio Oriente |
1/124 1/71 1/136 1/55 |
99% 99% 99% 99% |
1/12301 1/7001 1/13501 1/5401 |
1/6101296 1/1988284 1/7344544 1/1188220 |
| CAPN3 | AR | Distrofia Muscular de Cinturas Tipo 2A | Geral Europeu |
<1/500 1/103 |
98% 98% |
<1/24951 1/5101 |
<1/49902000 1/2101612 |
| CBS | AR | Homocistinúria Devida à Deficiência da Cistationina Beta-sintase | Geral Europeu Médio Oriente |
1/224 1/86 1/21 |
99% 99% 99% |
1/22301 1/8501 1/2001 |
1/19981696 1/2924344 1/168084 |
| CDH23 | AR, DG | Síndrome de Usher tipo 1D | Geral | 1/285 | 90% | 1/2841 | 1/3238740 |
| CEP290 | AR | Síndrome de Bardet-Biedl 14 (+) | Geral | 1/190 | 98% | 1/9451 | 1/7182760 |
| CEP290 | AR | Síndrome de Joubert 5 (+) | Geral | 1/190 | 98% | 1/9451 | 1/7182760 |
| CEP290 | AR | Amaurose Congénita de Leber 10 (+) | Geral | 1/190 | 98% | 1/9451 | 1/7182760 |
| CEP290 | AR | Síndrome de Meckel 4 (+) | Geral | 1/190 | 98% | 1/9451 | 1/7182760 |
| CEP290 | AR | Síndrome Senior-Loken 6 (+) | Geral | 1/190 | 98% | 1/9451 | 1/7182760 |
| CEP290 | AR | Doenças Relacionadas com CEP290 | Geral | 1/190 | 98% | 1/9451 | 1/7182760 |
| CERKL | AR | Retinite Pigmentosa 26 | Geral | 1/480 | 98% | 1/7351 | 1/4351792 |
| CFTR | AR | Fibrose Quística | Geral Afro-americano Africano Judeu Ashkenazi Europeu Este da Ásia Latino |
1/32 1/61 1/61 1/24 1/25 1/94 1/58 |
99% 99% 99% 99% 99% 99% 99% |
1/3101 1/6001 1/6001 1/2301 1/2401 1/9301 1/5701 |
1/396928 1/1464244 1/1464244 1/220896 1/240100 1/3497176 1/1322632 |
| CHM | XL | Coroideremia | Geral | 1/25000 | 95% | 1/499981 | 1/1999924 |
| CHRNE | AR | Síndrome Miasténico Congénito, Relacionado com CHRNE | Geral | 1/408 | 99% | 1/40701 | 1/66424032 |
| CHRNG | AR | Síndrome de Pterígio Múltiplo | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| CIITA | AR | Síndrome do Linfócito Nu: Tipo II | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| CLN5 | AR | Lipofuscinose Ceroide Neuronal: Relacionada com CLN5 | Geral Finlandês |
<1/500 1/115 |
95% 95% |
<1/9981 1/2281 |
<1/19962000 1/1049260 |
| CLN6 | AR | Lipofuscinose Ceroide Neuronal: Relacionada com CLN6 | Geral | <1/500 | 92% | <1/6239 | <1/12478000 |
| CLN8 | AR | Lipofuscinose Ceroide Neuronal: Relacionada com CLN8 | Geral Finlandês |
<1/500 1/135 |
95% 95% |
<1/9981 1/2681 |
<1/19962000 1/1447740 |
| CLRN1 | AR | Síndrome de Usher Tipo 3A | Geral Judeu Ashkenazi Finlandês |
1/500 1/120 1/70 |
98% 98% 98% |
1/24951 1/5951 1/3451 |
1/49902000 1/2856480 1/966280 |
| COL4A3 | AR, DG | Síndrome de Alporte, relacionado com COL4A3 | Geral Judeu Ashkenazi |
1/267 1/188 |
98% 98% |
1/13301 1/9351 |
1/14205468 1/7031952 |
| COL4A4 | AR, DG | Síndrome de Alporte, relacionado com COL4A4 | Geral | 1/267 | 98% | 1/13301 | 1/14205468 |
| COL4A5 | XL | Síndrome de Alporte, relacionado com COL4A5 | Geral | 1/139 | 98% | 1/6901 | 1/27604 |
| COL7A1 | AR | Epidermólise Bolhosa Distrófica Recessiva | Geral | 1/196 | 97% | 1/6501 | 1/5096784 |
| CPT1A | AR | Deficiência de Carnitina-Palmitoil Transferase: Tipo IA | Geral Huterita |
1/354 1/16 |
90% 90% |
1/3531 1/151 |
1/4999896 1/9664 |
| CPT2 | AR | Deficiência de Carnitina-Palmitoil Transferase: Tipo II | Geral Judeu Ashkenazi |
<1/500 1/51 |
95% 95% |
<1/9981 1/1001 |
<1/19962000 1/204204 |
| CTNS | AR | Cistinose | Geral Britânico |
1/158 1/81 |
99% 99% |
1/15701 1/8001 |
1/9923032 1/2592324 |
| CTSC | AR | Síndrome de Papillon-Lefèvre | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| CTSK | AR | Picnodisostose | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| CYBA | AR | Doença Granulomatosa Crónica | Geral | 1/224 | 99% | 1/22301 | 1/19981696 |
| CYBB | XL | Doença Granulomatosa Crónica, ligado ao X | Geral | 1/149254 | 99% | 1/14925301 | 1/59701204 |
| CYP11B1 | AR | Hiperplasia Congénita da Suprarrenal por Défice de 17-Beta-Hidroxilase | Geral Judeu Marroquino |
1/158 1/35 |
98% 98% |
1/7851 1/1701 |
1/4961832 1/238140 |
| CYP11B2 | AR | Deficiência de Corticosterona Metiloxidase | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| CYP17A1 | AR | Hiperplasia Congénita da Suprarrenal por Défice de 17-Alfa-Hidroxilase | Geral | 1/500 | 98% | 1/24951 | <1/10000000 |
| CYP19A1 | AR | Deficiência de Aromatase | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| CYP1B1 | AR | Glaucoma Congénito Primário | Geral | 1/50 | 99% | 1/4901 | 1/980200 |
| CYP21A2 | AR | Hiperplasia Congénita da Suprarrenal por Défice de 21 - Hidroxilase | Geral Inuíte Médio Oriente |
1/61 1/9 1/35 |
99% 99% 99% |
1/6001 1/801 1/3401 |
1/1464244 1/28836 1/476140 |
| CYP27A1 | AR | Xantomatose Cerebrotendinosa | Geral Judeu Marroquino |
1/500 1/5 |
98% 98% |
1/24951 1/201 |
1/49902000 1/4020 |
| DBT | AR | Doença da Urina de Xarope de Bordo Tipo II | Geral | 1/481 | 98% | 1/24001 | 1/46177924 |
| DCLRE1C | AR | Imunodeficiência Combinada Grave com Sensibilidade à Radiação Ionizante | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| DHCR7 | AR | Síndrome de Smith-Lemli-Opitz | Geral Afro-americano Africano Judeu Ashkenazi |
1/30 1/138 1/138 1/36 |
96% 96% 96% 96% |
1/726 1/3426 1/3426 1/876 |
1/87120 1/1891152 1/1891152 1/126144 |
| DHDDS | AR | Retinite Pigmentosa 59 | Geral Judeu Ashkenazi |
1/296 1/118 |
98% 98% |
1/14751 1/5851 |
1/17465184 1/2761672 |
| DLD | AR | Deficiência de Dihidrolipoamida Desidrogenase | Geral Judeu Ashkenazi |
1/500 1/107 |
98% 98% |
1/24951 1/5301 |
1/49902000 1/2268828 |
| DMD | XL | Distrofia Muscular de Duchenne (+) | Geral | 1/2350 | 93% | 1/33558 | 1/134232 |
| DMD | XL | Distrofias Musculares Relacionadas com DMD | Geral | 1/2350 | 93% | 1/33558 | 1/315445200 |
| DNAI1 | AR | Discinesia Ciliar Primária: Relacionada com DNAI1 | Geral | 1/230 | 98% | 1/11451 | 1/10534920 |
| DNAI2 | AR | Discinesia Ciliar Primária: Relacionada com DNAI2 | Geral | 1/447 | 98% | 1/22301 | 1/39874188 |
| DOK7 | AR | Síndrome Miasténico Congénito: Relacionado com DOK7 | Geral | 1/472 | 98% | 1/23551 | 1/44464288 |
| DYSF | AR | Distrofia Muscular de Cinturas: Tipo 2B | Geral Japonês Judeu Líbio |
<1/500 1/332 1/18 |
95% 95% 95% |
1/9981 1/6621 1/341 |
<1/19962000 1/8792688 1/24552 |
| EDA | XL | Displasia Ectodérmica Hipohidrótica: Ligada ao X | Geral | 1/14167 | 99% | 1/1416601 | 1/5666404 |
| EIF2AK3 | AR | Síndrome de Wolcott-Rallison | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| EIF2B5 | AR | Leucoencefalopatia com Desaparecimento da Substância Branca | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| EMD | XL | Miopatia de Emery-Dreifuss: Ligado ao X | Geral | 1/81967 | 99% | 1/8196601 | 1/32786404 |
| ERCC6 | AR | Síndrome de Cockayne Tipo B (+) | Geral Japonês |
1/500 1/74 |
99% 99% |
1/49901 1/7301 |
1/99802000 1/2161096 |
| ERCC6 | AR | Síndrome de Sanctis-Cacchione (+) | Geral Japonês |
1/500 1/74 |
99% 99% |
1/49901 1/7301 |
1/99802000 1/2161096 |
| ERCC6 | AR | Doenças Relacionadas com ERCC6 | Geral Japonês |
1/500 1/74 |
99% 99% |
1/49901 1/7301 |
1/99802000 1/2161096 |
| ERCC8 | AR | Síndrome de Cockayne Tipo A | Geral | 1/820 | 98% | 1/41051 | 1/134975688 |
| ETFA | AR | Acidúria Glutárica Tipo IIA | Geral | 1/500 | 98% | 1/24951 | 1/49902000 |
| ETFB | AR | Acidúria Glutárica Tipo IIB | Geral | 1/500 | 98% | 1/24951 | 1/49902000 |
| ETFDH | AR | Acidúria Glutárica Tipo IIC | Geral Este da Ásia |
1/250 1/74 |
98% 98% |
1/12451 1/3651 |
1/12451000 1/1080696 |
| ETHE1 | AR | Acidúria Etilmalónica | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| EVC | AR | Síndrome de Ellis-van Creveld, Relacionado com EVC (+) | Geral Amish |
1/142 1/7 |
98% 98% |
1/7051 1/301 |
1/4004968 1/8428 |
| EVC | AR | Disostose Acrofacial Tipo Weyers, Relacionada com EVC (+) | Geral Amish |
1/142 1/7 |
98% 98% |
1/7051 1/301 |
1/4004968 1/8428 |
| EVC | AR | Doenças Relacionadas com EVC | Geral Amish |
1/142 1/7 |
98% 98% |
1/7051 1/301 |
1/4004968 1/8428 |
| EVC2 | AR | Síndrome de Ellis-van Creveld, Relacionado com EVC2 (+) | Geral Amish |
1/240 1/7 |
98% 98% |
1/11951 1/301 |
1/11472960 1/8428 |
| EVC2 | AR | Disostose Acrodental Tipo Weyers, Relacionada com EVC2 (+) | Geral Amish |
1/240 1/7 |
98% 98% |
1/11951 1/301 |
1/11472960 1/8428 |
| EVC2 | AR | Doenças Relacionadas com EVC2 | Geral Amish |
1/240 1/7 |
98% 98% |
1/11951 1/301 |
1/11472960 1/8428 |
| EXOSC3 | AR | Hipoplasia Ponto-Cerebelar: Relacionada com EXOSC3 | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| F8 | XL | Hemofilia A | Geral | 1/3250 | 48% | 1/6249 | 1/24996 |
| F9 | XL | Hemofilia B | Geral | 1/15000 | 99% | 1/1499901 | 1/5999604 |
| FAH | AR | Tirosinemia, Tipo I | Geral Judeu Ashkenazi Finlandês Franco-Canadense Sul da Ásia / Índia |
1/99 1/150 1/122 1/66 1/172 |
95% 95% 95% 95% 95% |
1/1961 1/2981 1/2421 1/1301 1/3421 |
1/776556 1/1788600 1/1181448 1/343464 1/2353648 |
| FAM161A | AR | Retinite Pigmentosa 28 | Geral | 1/296 | 98% | 1/14751 | 1/17465184 |
| FANCA | AR | Anemia de Fanconi Grupo A | Geral | 1/239 | 98% | 1/11901 | 1/11377356 |
| FANCC | AR | Anemia de Fanconi: Tipo C | Geral Judeu Ashkenazi |
1/535 1/99 |
99% 99% |
1/53401 1/9801 |
1/114278140 1/3881196 |
| FANCG | AR | Anemia de Fanconi: Tipo G | Geral | 1/632 | 90% | 1/6311 | 1/15954208 |
| FH | AR | Deficiência de Fumarase | Geral | <1/500 | 90% | <1/4991 | <1/9982000 |
| FKRP | AR | Distrofia-Distroglicanopatia Muscular Relacionada com FKRP | Geral | 1/158 | 98% | 1/7851 | 1/4961832 |
| FKTN | AR | Doenças Relacionadas com FKTN | Geral Judeu Ashkenazi Japonês |
<1/500 1/150 1/82 |
99% 99% 99% |
<1/49901 1/14901 1/8101 |
<1/99802000 1/8940600 1/2657128 |
| FKTN | AR | Distrofia Muscular Congénita Tipo Fukuyama (+) | Geral Judeu Ashkenazi Japonês |
<1/500 1/150 1/82 |
99% 99% 99% |
1/49901 1/14901 1/8101 |
<1/99802000 1/8940600 1/2657128 |
| FKTN | AR | Distrofia Muscular Devida a uma Distroglicanopatia, Relacionada com FKTN (+) | Geral Judeu Ashkenazi Japonês |
<1/500 1/150 1/82 |
99% 99% 99% |
<1/49901 1/14901 1/8101 |
<1/99802000 1/8940600 1/2657128 |
| FMR1 | XL | Síndrome de X Frágil | Geral Judeu Ashkenazi |
1/259 1/115 |
99% 99% |
1/25801 1/11401 |
1/103204 1/45604 |
| G6PC | AR | Doença do Armazenamento do Glicogénio Tipo 1a | Geral Judeu Ashkenazi |
1/177 1/64 |
95% 95% |
1/3521 1/1261 |
1/2492868 1/322816 |
| G6PD | XL | Deficiência de Glicose 6 Fosfato Desidrogenase | Geral | 1/7 | 98% | 1/301 | 1/1204 |
| GAA | AR | Doença de Pompe | Geral Afro-americano Africano Este da Ásia |
1/100 1/60 1/60 1/112 |
98% 98% 98% 98% |
1/4951 1/2951 1/2951 1/5551 |
1/1980400 1/708240 1/708240 1/2486848 |
| GALC | AR | Doença de Krabbe | Geral Druso Israel |
1/158 1/6 |
99% 99% |
1/15701 1/501 |
1/9923032 1/12024 |
| GALK1 | AR | Deficiência de Galactoquinase | Geral Irlandês |
1/110 1/64 |
95% 95% |
1/2181 1/1261 |
1/959640 1/322816 |
| GALNS | AR | Mucopolissacaridose Tipo IVA (Síndrome de Morquio: Tipo A) | Geral | 1/224 | 97% | 1/7434 | 1/6660864 |
| GALT | AR | Galactosémia | Geral Africano Afro-americano |
1/110 1/94 1/94 |
95% 95% 95% |
1/2181 1/1861 1/1861 |
1/959640 1/699736 1/699736 |
| GAMT | AR | Deficiência de Guanidinoacetato Metiltransferase | Geral | 1/371 | 99% | 1/37001 | 1/54909484 |
| GBA | AR | Doença de Gaucher | Geral Africano Afro-americano Judeu Ashkenazi |
1/77 1/35 1/35 1/15 |
99% 99% 99% 99% |
1/7601 1/3401 1/3401 1/1401 |
1/2341108 1/476140 1/476140 1/84060 |
| GBE1 | AR | Doença de Armazenamento de Glicogénio IV | Geral | 1/387 | 99% | 1/38601 | 1/59754348 |
| GCDH | AR | Acidúria Glutárica Tipo I | Geral Amish |
1/87 1/9 |
98% 98% |
1/4301 1/401 |
1/1496748 1/14436 |
| GDF5 | AR | Síndrome de Du Pan | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| GJB1 | XL | Doença de Charcot-Marie-Tooth com Surdez, Ligada ao X Tipo I | Geral | 1/667 | 90% | 1/6661 | 1/26644 |
| GJB2 | AR | Surdez não Sindrómica, Relacionada com GJB2 | Geral Africano Afro-americano Judeu Ashkenazi Europeu Latino Médio Oriente Sul da Ásia / Índia |
1/42 1/25 1/25 1/21 1/33 1/100 1/83 1/148 |
99% 99% 99% 99% 99% 99% 99% 99% |
1/4101 1/2401 1/2401 1/2001 1/3201 1/9901 1/8201 1/14701 |
1/688968 1/240100 1/240100 1/168084 1/422532 1/3960400 1/2722732 1/8702992 |
| GLA | XL | Doença de Fabry | Geral | 1/25000 | 99% | 1/2499901 | 1/9999604 |
| GLB1 | AR | Mucopolissacaridose Tipo IVB (Síndrome de Morquio: Tipo B) | Geral Maltês Romani |
1/134 1/30 1/50 |
99% 99% 99% |
1/13301 1/2901 1/4901 |
1/7129336 1/348120 1/980200 |
| GLB1 | AR | Gangliosidose GM1 (+) | Geral Maltês Romani |
1/134 1/30 1/50 |
99% 99% 99% |
1/13301 1/2901 1/4901 |
1/7129336 1/348120 1/980200 |
| GLB1 | AR | Doenças Relacionadas com GLB1 | Geral Maltês Romani |
1/134 1/30 1/50 |
99% 99% 99% |
1/13301 1/2901 1/4901 |
1/7129336 1/348120 1/980200 |
| GLDC | AR | Encefalopatia por Glicina: Relacionada com GLDC | Geral Canadense (British Columbia) Finlandês |
1/193 1/125 1/117 |
98% 99% 99% |
1/9601 1/12401 1/11601 |
1/7411972 1/6200500 1/5429268 |
| GNE | AR | Miopatia de Corpos de Inclusão: Tipo 2 (Miopatia de Nonaka) | Geral Judeu Iraniano |
<1/500 1/11 |
80% 80% |
<1/2496 1/51 |
<1/4992000 1/2244 |
| GNPTAB | AR | Mucolipidose II Alfa / Beta (+) | Geral | <1/500 | 95% | <1/9981 | <1/19962000 |
| GNPTAB | AR | Mucolipidose III Alfa / Beta (+) | Geral | <1/500 | 95% | <1/9981 | <1/19962000 |
| GNPTAB | AR | Doenças Relacionadas com GNPTAB | Geral | <1/500 | 95% | <1/9981 | <1/19962000 |
| GNS | AR | Mucopolissacaridose IIID (Síndrome de Sanfilippo D) | Geral | 1/500 | 98% | 1/24951 | 1/49902000 |
| GRHPR | AR | Hiperoxalúria Primária: Tipo II | Geral | <1/500 | 99% | 1/49901 | <1/99802000 |
| GUCY2D | AR | Amaurose Congénita de Leber 1: Relacionada a GUCY2D | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| GUSB | AR | Mucopolissacaridose Tipo VII (Síndrome de Sly) | Geral | 1/250 | 98% | 1/12451 | 1/12451000 |
| HADHA | AR | Deficiência de 3-Hidroxiacil-CoA-Deshidrogenase de Cadeia Longa (LCHAD) (+) | Geral Finlandês |
<1/500 1/124 |
98% 98% |
<1/24951 1/6151 |
<1/49902000 1/3050896 |
| HADHA | AR | Deficiência Proteína Trifuncional: Relacionada com HADHA (+) | Geral Finlandês |
<1/500 1/124 |
98% 98% |
<1/24951 1/6151 |
<1/49902000 1/3050896 |
| HADHA | AR | Doenças Relacionadas com HADHA | Geral Finlandês |
<1/500 1/124 |
98% 98% |
<1/24951 1/6151 |
<1/49902000 1/3050896 |
| HADHB | AR | Deficiência da Proteína Trifuncional Mitocondrial: Relacionada com HADHB | Geral Finlandês |
<1/500 1/124 |
98% 98% |
<1/24951 1/6151 |
<1/49902000 1/3050896 |
| HAX1 | AR | Neutropénia Congénita Severa: Relacionada com HAX1 | Geral | 1/224 | 98% | 1/11151 | 1/9991296 |
| HBA1 | AR, DG | Alfa Talassemia | Geral Africano Afro-americano Judeu Ashkenazi Este da Ásia Médio Oriente Sul da Ásia / Índia |
1/20 1/3 1/3 1/13 1/8 1/3 1/5 |
90% 90% 90% 90% 90% 90% 90% |
1/191 1/21 1/21 1/121 1/71 1/21 1/41 |
1/15280 1/252 1/252 1/6292 1/2272 1/252 1/820 |
| HBA2 | AR, DG | Alfa Talassemia | Geral Africano Afro-americano Judeu Ashkenazi Este da Ásia Médio Oriente Sul da Ásia / Índia |
1/20 1/3 1/3 1/13 1/8 1/3 1/5 |
90% 90% 90% 90% 90% 90% 90% |
1/191 1/21 1/21 1/121 1/71 1/21 1/41 |
1/15280 1/252 1/252 1/6292 1/2272 1/252 1/820 |
| HBB | AR | Beta Talassemia | Geral Africano Afro-americano Este da Ásia Latino Mediterrâneo Sul da Ásia / Índia |
1/158 1/10 1/10 1/50 1/128 1/3 1/25 |
95% 95% 95% 95% 95% 95% 95% |
1/3141 1/181 1/181 1/981 1/2541 1/41 1/481 |
1/1985112 1/7240 1/7240 1/196200 1/1300992 1/492 1/48100 |
| HBB | AR | Anemia de Células Falciformes | Geral Africano Afro-americano Este da Ásia Latino Mediterrâneo Sul da Ásia / Índia |
1/158 1/10 1/10 1/50 1/128 1/3 1/25 |
95% 95% 95% 95% 95% 95% 95% |
1/3141 1/181 1/181 1/981 1/2541 1/41 1/481 |
1/1985112 1/7240 1/7240 1/196200 1/1300992 1/492 1/48100 |
| HBB | AR | Hemoglobinopatia: HbC | Geral Africano Afro-americano Este da Ásia Latino Mediterrâneo Sul da Ásia / Índia |
1/158 1/10 1/10 1/50 1/128 1/3 1/25 |
95% 95% 95% 95% 95% 95% 95% |
1/3141 1/181 1/181 1/981 1/2541 1/41 1/481 |
1/1985112 1/7240 1/7240 1/196200 1/1300992 1/492 1/48100 |
| HBB | AR | Doenças Relacionadas com HBB | Geral Africano Afro-americano Este da Ásia Latino Mediterrâneo Sul da Ásia / Índia |
1/158 1/10 1/10 1/50 1/128 1/3 1/25 |
95% 95% 95% 95% 95% 95% 95% |
1/3141 1/181 1/181 1/981 1/2541 1/41 1/481 |
1/1985112 1/7240 1/7240 1/196200 1/1300992 1/492 1/48100 |
| HEXA | AR | Doença de Tay-Sachs | Geral Judeu Ashkenazi |
1/300 1/27 |
99% 99% |
1/29901 1/2601 |
1/35881200 1/280908 |
| HEXB | AR | Doença Sandhoff | Geral | 1/600 | 98% | 1/29951 | 1/71882400 |
| HFE2 | AR | Hemocromatose: Tipo 2A | Geral | 1/500 | 99% | 1/49901 | 1/99802000 |
| HGD | AR | Alcaptonúria | Geral | 1/250 | 90% | 1/2491 | 1/2491000 |
| HGSNAT | AR | Mucopolissacaridose Tipo IIIC (Síndrome de Sanfilippo C) | Geral Europeu |
1/434 1/345 |
98% 98% |
1/21651 1/17201 |
1/37586136 1/23737380 |
| HLCS | AR | Deficiência de Holocarboxilase Sintetase | Geral | 1/500 | 98% | 1/24951 | 1/49902000 |
| HMGCL | AR | Deficiência de 3-Hidroxi-3-metilglutaril-CoA Liase | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| HOGA1 | AR | Hiperoxalúria Primária Tipo III | Geral | 1/184 | 99% | 1/18301 | 1/13469536 |
| HPS1 | AR | Síndrome de Hermansky-Pudlak 1 | Geral Porto-Riquenho |
1/354 1/21 |
98% 98% |
1/17651 1/1001 |
1/24993816 1/84084 |
| HPS3 | AR | Síndrome de Hermansky-Pudlak 3 | Geral | 1/354 | 98% | 1/17651 | 1/24993816 |
| HPS4 | AR | Síndrome de Hermansky-Pudlak 4 | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| HSD17B3 | AR | Deficiência de 17 - Beta Hidroxiesteróide Desidrogenase | Geral Palestino |
1/192 1/8 |
98% 98% |
1/9551 1/351 |
1/7335168 1/11232 |
| HSD17B4 | AR | Deficiência de Proteína D-Bifuncional | Geral | 1/158 | 98% | 1/7851 | 1/4961832 |
| HSD3B2 | AR | Hiperplasia Congénita Adrenal por Deficiência de 3-Beta-Hidroxiesteróide Desidrogenase 2 | Geral | <1/500 | 98% | 1/24951 | <1/10000000 |
| IDS | XL | Mucopolissacaridose Tipo II (Síndrome de Hunter) | Geral | 1/50000 | 91% | 1/555545 | 1/2222180 |
| IDUA | AR | Mucopolissacaridose Tipo I (Síndrome de Hurler) | Geral Europeu |
<1/500 1/153 |
95% 95% |
<1/9981 1/3041 |
<1/19962000 1/1861092 |
| KBKAP | AR | Disautonomia Familiar | Geral Judeu Ashkenazi |
1/300 1/31 |
99% 99% |
1/29901 1/3001 |
1/35881200 1/372124 |
| IL2RG | XL | Imunodeficiência Combinada Severa: Ligada ao X | Geral | 1/25000 | 99% | 1/2499901 | 1/9999604 |
| IVD | AR | Acidemia Isovalérica | Geral Africano Afro-americano Europeu Este da Ásia |
1/167 1/100 1/100 1/115 1/407 |
90% 90% 90% 90% 90% |
1/1661 1/991 1/991 1/1141 1/4061 |
1/1109548 1/396400 1/396400 1/524860 1/6611308 |
| KCNJ11 | AR | Diabetes Mellitus Neonatal Permanente (+) | Geral Europeu |
1/423 1/232 |
99% 99% |
1/42201 1/23101 |
1/71404092 1/21437728 |
| KCNJ11 | AR | Hiperinsulinismo Familiar Tipo 2: Relacionado com KCNJ11 (+) | Geral Europeu |
1/423 1/232 |
99% 99% |
1/42201 1/23101 |
1/71404092 1/21437728 |
| KCNJ11 | AR | Doenças Relacionadas com KCNJ11 | Geral Europeu |
1/423 1/232 |
99% 99% |
1/42201 1/23101 |
1/71404092 1/21437728 |
| LAMA3 | AR | Epidermólise Bolhosa Juncional de Herlitz: Relacionada com LAMA3 (+) | Geral | 1/781 | 98% | 1/39001 | 1/121839124 |
| LAMA3 | AR | Síndrome Laringo-Onico-Cutânea (+) | Geral | 1/781 | 98% | 1/39001 | 1/121839124 |
| LAMA3 | AR | Doenças Relacionadas com LAMA3 | Geral | 1/781 | 98% | 1/39001 | 1/121839124 |
| LAMB3 | AR | Epidermólise Bolhosa: Relacionada com LAMB3 | Geral | 1/781 | 98% | 1/39001 | 1/121839124 |
| LAMC2 | AR | Epidermólise Bolhosa: Relacionada com LAMC2 | Geral | 1/781 | 98% | 1/39001 | 1/121839124 |
| LCA5 | AR | Amaurose Congénita de Leber 5 | Geral | 1/500 | 98% | 1/24951 | 1/49902000 |
| LHCGR | AR | Síndrome de Hipoplasia de células de Leydig (Resistência à Hormona Luteinizante) | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| LIFR | AR | Síndrome de Stuve-Wiedemann | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| LIPA | AR | Deficiência da Lipase Ácida Lisossomal | Geral Europeu |
<1/500 1/112 |
99% 99% |
<1/49901 1/11101 |
<1/99802000 1/4973248 |
| LOXHD1 | AR | Perda Auditiva não Sindrómica, Relacionada com LOXHD1 | Geral Judeu Ashkenazi |
1/500 1/180 |
98% 98% |
1/24951 1/8951 |
1/49902000 1/6444720 |
| LPL | AR | Deficiência Familiar de Lipoproteína Lipase | Geral Franco-Canadense |
1/500 1/46 |
99% 99% |
1/49901 1/4501 |
1/99802000 1/828184 |
| LRPPRC | AR | Síndrome de Leigh com Deficiência de Complexo IV | Geral Feroês Franco-Canadense |
1/447 1/21 1/22 |
98% 98% 98% |
1/22301 1/1001 1/1051 |
1/39874188 1/84084 1/92488 |
| LYST | AR | Síndrome de Chediak-Higashi | Geral | <1/500 | 90% | <1/4991 | <1/9982000 |
| MAN2B1 | AR | Alfa-Manosidose | Geral Europeu |
1/354 1/274 |
99% 99% |
1/35301 1/27301 |
1/49986216 1/29921896 |
| MCCC1 | AR | Deficiência de 3-Metilcrotonil-CoA Carboxilase 1 (Deficiência de 3-MCC) | Geral | 1/95 | 98% | 1/4701 | 1/1786380 |
| MCCC2 | AR | Deficiência de 3-Metilcrotonil-CoA Carboxilase 2 (Deficiência de 3-MCC) | Geral | 1/95 | 98% | 1/4701 | 1/1786380 |
| MCOLN1 | AR | Mucolipidose IV | Geral Judeu Ashkenazi |
1/300 1/100 |
99% 99% |
1/29901 1/9901 |
1/35881200 1/3960400 |
| MED17 | AR | Atrofia Cerebral e Cerebelosa Infantil com Microcefalia Progressiva Pós Natal | Geral | <1/500 | 99% | <1/49901 | <1/99802000 |
| MEFV | AR | Febre Mediterrânea Familiar | Geral Mediterrâneo |
1/20 1/7 |
99% 90% |
1/1901 1/61 |
1/152080 1/1708 |
| MFSD8 | AR | Lipofuscinose Neuronal Ceróide: Relacionada com MFSD8 | Geral | <1/500 | 95% | <1/9981 | <1/19962000 |
| MKS1 | AR | Síndrome de Meckel: Tipo 1 (+) | Geral Finlandês |
1/260 1/47 |
98% 98% |
1/12951 1/2301 |
1/13469040 1/432588 |
| MKS1 | AR | Síndrome de Bardet-Biedl 13 (+) | Geral Finlandês |
1/260 1/47 |
98% 98% |
1/12951 1/2301 |
1/13469040 1/432588 |
| MKS1 | AR | Síndrome de Joubert 28 (+) | Geral Finlandês |
1/260 1/47 |
98% 98% |
1/12951 1/2301 |
1/13469040 1/432588 |
| MKS1 | AR | Doenças Relacionadas com MKS1 | Geral Finlandês |
1/260 1/47 |
98% 98% |
1/12951 1/2301 |
1/13469040 1/432588 |
| MLC1 | AR | Leucoencefalopatia Megalencefálica com Quistos Subcorticais | Geral | <1/500 | 97% | <1/16634 | <1/33268000 |
| MLYCD | AR | Deficiência de Malonil-CoA Descarboxilase | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| MMAA | AR | Acidemia Metilmalónica Tipo cbIA | Geral | 1/301 | 97% | 1/10001 | 1/12041204 |
| MMAB | AR | Acidemia Metilmalónica Tipo cbIB | Geral | 1/435 | 98% | 1/21701 | 1/37759740 |
| MMACHC | AR | Acidúria Metilmalónica e Homocistinúria, Tipo cbIC | Geral | 1/134 | 90% | 1/1331 | 1/713416 |
| MPI | AR | Defeitos Congénitos da Glicosilação: Tipo Ib | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| MPL | AR | Trombocitopenia Amegacariocítica Congénita | Geral Judeu Ashkenazi |
1/102 1/55 |
98% 98% |
1/5051 1/2701 |
1/2060808 1/594220 |
| MPV17 | AR | Síndrome de Depleção do DNA Mitocondrial Hepatocerebral, Relacionado com MPV17 | Geral Nativo Americano |
<1/500 1/20 |
96% 96% |
1/12476 1/476 |
<1/24952000 1/38080 |
| MTM1 | XL | Miopatia Miotubular: Ligada ao X | Geral | 1/25000 | 98% | 1/1249951 | 1/4999804 |
| MTTP | AR | Abetalipoproteinemia | Geral Judeu Ashkenazi |
<1/500 1/180 |
98% 98% |
<1/24951 1/8951 |
<1/49902000 1/6444720 |
| MUT | AR | Acidemia Metilmalónica, Relacionada com MUT | Geral Este da Ásia Médio Oriente |
1/195 1/53 1/52 |
96% 96% 96% |
1/4851 1/1301 1/1276 |
1/3783780 1/275812 1/265408 |
| MYO15A | AR | Surdez não Sindrómica Relacionada com MYO15A | Geral Balinês Paquistanês |
1/500 1/6 1/77 |
98% 98% 98% |
1/24951 1/251 1/3801 |
1/49902000 1/6024 1/1170708 |
| MYO7A | AR | Surdez não Sindrómica Relacionada com MYO7A (+) | Geral Este da Ásia |
1/206 1/62 |
98% 98% |
1/10521 1/3051 |
1/8669304 1/756648 |
| MYO7A | AR | Síndrome de Usher tipo 1B (+) | Geral Este da Ásia |
1/206 1/62 |
98% 98% |
1/10521 1/3051 |
1/8446824 1/756648 |
| MYO7A | AR | Doenças Relacionadas com MYO7A | Geral Este da Ásia |
1/206 1/62 |
98% 98% |
1/10521 1/3051 |
1/8446824 1/756648 |
| NAGLU | AR | Mucopolissacaridose Tipo IIIB (Síndrome de Sanfilippo B) | Geral Europeu Este da Ásia |
<1/500 1/346 1/298 |
99% 99% 99% |
<1/49901 1/34501 1/29701 |
<1/99802000 1/47749384 1/35403592 |
| NBN | AR | Síndrome de Rotura de Nijmegen | Geral | 1/158 | 99% | 1/15701 | 1/9923032 |
| NDUFS6 | AR | Deficiência do Complexo I Mitocondrial: Relacionada com NDUFS6 (Síndrome de Leigh) | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| NEB | AR | Miopatia Nemalítica | Geral Amish Judeu Ashkenazi Finlandês |
1/112 1/11 1/108 1/112 |
98% 98% 98% 98% |
1/5551 1/501 1/5351 1/5551 |
1/2486848 1/22044 1/2311632 1/2486848 |
| NPC1 | AR | Doença de Niemann-Pick, Tipo C1 | Geral | 1/194 | 90% | 1/1931 | 1/1498456 |
| NPC2 | AR | Doença de Niemann-Pick, Tipo C2 | Geral | 1/194 | 99% | 1/19301 | 1/14977576 |
| NPHS1 | AR, DG | Síndrome Nefrótico Congénito Tipo 1 | Geral Finlandês |
1/289 1/50 |
98% 98% |
1/14401 1/2451 |
1/16647556 1/490200 |
| NPHS2 | AR, DG | Síndrome Nefrótico Congénito Tipo 2 | Geral Finlandês |
1/289 1/50 |
98% 98% |
1/14401 1/2451 |
1/16647556 1/490200 |
| NR2E3 | AR | Síndrome de Distrofia Vitreorretiniana (+) | Geral | 1/209 | 98% | 1/10401 | 1/8695236 |
| NR2E3 | AR | Retinite Pigmentosa 37 (+) | Geral | 1/209 | 98% | 1/10401 | 1/8695236 |
| NR2E3 | AR | Condições Relacionadas com NR2E3 | Geral | 1/209 | 98% | 1/10401 | 1/8695236 |
| NTRK1 | AR | Insensibilidade Congénita à Dor com Anidrose | Geral | <1/500 | 99% | <1/49901 | <1/99802000 |
| OCRL | XL | Doença de Dent 2 (+) | Geral | <1/250000 | 95% | <1/4999981 | <1/19999924 |
| OCRL | XL | Síndrome de Lowe (+) | Geral | 1/250000 | 95% | 1/4999981 | 1/19999924 |
| OCRL | XL | Doenças Relacionadas com OCRL | Geral | 1/250000 | 95% | 1/4999981 | 1/19999924 |
| OPA3 | AR | Síndrome de Costeff | Geral Judeu Iraquiano |
<1/500 1/50 |
98% 98% |
<1/24951 1/2451 |
<1/10000000 1/490200 |
| OTC | XL | Deficiência de Ornitina Transcarbamilase | Geral | 1/7000 | 90% | 1/69991 | 1/279964 |
| PAH | AR | Deficiência de Fenilalanina Hidroxilase (Fenilcetonúria) | Geral Europeu Médio Oriente Sudeste Asiático |
1/93 1/63 1/74 1/59 |
99% 99% 99% 99% |
1/9201 1/6201 1/7301 1/5801 |
1/3422772 1/1562652 1/2161096 1/1369036 |
| PC | AR | Deficiência de Piruvato Carboxilase | Geral | 1/250 | 95% | 1/4981 | 1/4981000 |
| PCCA | AR | Acidemia Propiónica: Relacionada com PCCA | Geral Nativo Americano |
1/224 1/85 |
96% 96% |
1/5576 1/2101 |
1/4996096 1/714340 |
| PCCB | AR | Acidemia Propiónica: Relacionada com PCCB | Geral Nativo Americano |
1/224 1/85 |
99% 99% |
1/22301 1/8401 |
1/19981696 1/2856340 |
| PCDH15 | AR, DG | Surdez não Sindrómica Relacionada com PCDH15 (+) | Geral Judeu Ashkenazi |
1/395 1/72 |
98% 98% |
1/19701 1/3551 |
1/31127580 1/1022688 |
| PCDH15 | AR, DG | Síndrome de Usher Tipo 1F (+) | Geral Judeu Ashkenazi |
1/395 1/72 |
98% 98% |
1/19701 1/3551 |
1/31127580 1/1022688 |
| PCDH15 | AR, DG | Doenças Relacionadas com PCDH15 | Geral Judeu Ashkenazi |
1/395 1/72 |
98% 98% |
1/19701 1/3551 |
1/31127580 1/1022688 |
| PDHA1 | XL | Deficiência de Piruvato Desidrogenase, Deficiência E1-alfa: Ligado ao X | Geral | <1/250000 | 98% | <1/12499951 | <1/49999804 |
| PDHB | AR | Deficiência de Piruvato Desidrogenase, Deficiência E1-beta | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| PEX1 | AR | Síndrome de Zellweger, Relacionado com PEX1 | Geral | 1/147 | 95% | 1/2921 | 1/1717548 |
| PEX10 | AR | Síndrome de Zellweger, Relacionado com PEX10 | Geral Japonês |
1/500 1/354 |
95% 95% |
1/9981 1/7061 |
1/19962000 1/9998376 |
| PEX2 | AR | Síndrome de Zellweger, Relacionado com PEX2 | Geral Judeu Ashkenazi |
1/500 1/123 |
95% 95% |
1/9981 1/2441 |
1/19962000 1/1200972 |
| PEX6 | AR | Síndrome de Zellweger, Relacionado com PEX6 | Geral | 1/280 | 95% | 1/5581 | 1/6250720 |
| PEX7 | AR | Condrodisplasia Rizomélica Punctata: Tipo 1 | Geral | 1/158 | 99% | 1/15701 | 1/9923032 |
| PFKM | AR | Doença de Armazenamento de Glicogénio: Tipo VII | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| PHGDH | AR | Deficiência de Fosfoglicerato Desidrogenase | Geral Judeu Ashkenazi |
<1/500 1/280 |
98% 98% |
<1/24951 1/13951 |
<1/49902000 1/15625120 |
| PKHD1 | AR | Poliquistose Renal Associada a PKHD1 | Geral Judeu Ashkenazi |
1/70 1/107 |
98% 98% |
1/3451 1/5301 |
1/966280 1/2268828 |
| PMM2 | AR | Transtorno Congénito de Glicosilação Tipo 1a | Geral Judeu Ashkenazi Europeu |
<1/500 1/57 1/71 |
99% 99% 99% |
<1/49901 1/5601 1/7001 |
<1/99802000 1/1277028 1/1988284 |
| POLG | AR | Síndrome de Alpers-Huttenlocher (+) | Geral | 1/113 | 95% | 1/2241 | 1/1012932 |
| POLG | AR | Oftalmoplegia Externa Progressiva (+) | Geral | 1/113 | 95% | 1/2241 | 1/1012932 |
| POLG | AR | Espectro de Ataxia Neuropatía (+) | Geral | 1/113 | 95% | 1/2241 | 1/1012932 |
| POLG | AR | Transtornos Relacionados com POLG | Geral | 1/113 | 95% | 1/2241 | 1/1012932 |
| POLG | AR | Síndrome de Encefalomiopatia Neurogastrointestinal Mitocondrial (+) | Geral | 1/113 | 95% | 1/2241 | 1/1012932 |
| POMGNT1 | AR | Doença do Músculo-Olho-Cérebro (+) | Geral Finlandês |
1/462 1/111 |
98% 98% |
1/23051 1/5501 |
1/42598248 1/2442444 |
| POMGNT1 | AR | Retinite Pigmentosa 76 (+) | Geral Finlandês |
1/462 1/111 |
98% 98% |
1/23051 1/5501 |
1/42598248 1/2442444 |
| POMGNT1 | AR | Transtornos Relacionados com POMGNT1 | Geral Finlandês |
1/462 1/111 |
98% 98% |
1/23051 1/5501 |
1/42598248 1/2442444 |
| POR | AR | Síndrome de Antley-Bixler | Geral | 1/159 | 98% | 1/7901 | 1/5025036 |
| PPT1 | AR | Lipofuscinose Ceróide Neuronal, Relacionada com PPT1 | Geral Europeu Finlandês |
1/368 1/488 1/75 |
98% 98% 98% |
1/18351 1/24351 1/3701 |
1/27012672 1/47533152 1/1110300 |
| PROP1 | AR | Deficiência Combinada de Hormonas Pituitárias 2 | Geral | 1/45 | 98% | 1/2201 | 1/396180 |
| PRPS1 | XL | Síndrome de Arts (+) | Geral | <1/250000 | 98% | <1/12499951 | <1/49999804 |
| PRPS1 | XL | Doença de Charcot-Marie-Tooth com Surdez, Ligada ao X: Relacionada com PRPS1 (+) | Geral | <1/250000 | 98% | <1/12499951 | <1/49999804 |
| PRPS1 | XL | Super Estimulação da Fosforibosilpirofosfato Sintetase | Geral | <1/250000 | 98% | <1/12499951 | <1/49999804 |
| PRPS1 | XL | Síndrome de Rosenberg-Chutorian (+) | Geral | <1/250000 | 98% | <1/12499951 | <1/49999804 |
| PRPS1 | XL | Doenças Relacionadas com PRPS1 | Geral | <1/250000 | 98% | <1/12499951 | <1/49999804 |
| PTS | AR | Deficiência de Tetrahidrobiopterina | Geral | 1/354 | 96% | 1/8826 | <1/10000000 |
| PUS1 | AR | Miopatia Mitocondrial e Anemia Sideroblástica | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| PYGM | AR | Doença por Armazenamento de Glicogénio Tipo V | Geral Europeu |
<1/500 1/206 |
99% 99% |
<1/49901 1/20501 |
<1/99802000 1/16892824 |
| RAB23 | AR | Síndrome de Carpenter | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| RAG2 | AR | Síndrome de Omenn: Relacionado com RAG2 | Geral | 1/137 | 98% | 1/6801 | 1/3726948 |
| RAPSN | AR | Síndrome Miasténico Congénito, Relacionado com RAPSN (+) | Geral | <1/500 | 99% | <1/49901 | <1/99802000 |
| RAPSN | AR | Sequência Deformativa de Acinésia Fetal (+) | Geral | <1/500 | 99% | <1/49901 | <1/99802000 |
| RAPSN | AR | Doenças Relacionadas com RAPSN | Geral | <1/500 | 99% | <1/49901 | <1/99802000 |
| RARS2 | AR | Hipoplasia Ponto-Cerebelar Tipo 6 | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| RDH12 | AR | Amaurose Congénita de Leber: Tipo 13 | Geral Europeu |
<1/500 1/456 |
98% 98% |
<1/24951 1/22751 |
<1/49902000 1/41497824 |
| RLBP1 | AR | Distrofia da Retina: Relacionada com RLBP1 | Geral Europeu |
1/296 1/84 |
98% 98% |
1/14751 1/4151 |
1/17465184 1/1394736 |
| RMRP | AR | Displasia Anauxética (+) | Geral Amish Finlandês |
<1/500 1/16 1/76 |
99% 99% 99% |
<1/49901 1/1501 1/7501 |
<1/99802000 1/96064 1/2280304 |
| RMRP | AR | Hipoplasia de Cartilagem-Cabelo (+) | Geral Amish Finlandês |
<1/500 1/16 1/76 |
99% 99% 99% |
<1/49901 1/1501 1/7501 |
<1/99802000 1/96064 1/2280304 |
| RMRP | AR | Displasia Metafisária sem Hipotricose (+) | Geral Amish Finlandês |
<1/500 1/16 1/76 |
99% 99% 99% |
<1/49901 1/1501 1/7501 |
<1/99802000 1/96064 1/2280304 |
| RMRP | AR | Condições Relacionadas com RMRP | Geral Amish Finlandês |
<1/500 1/16 1/76 |
99% 99% 99% |
1/49901 1/1501 1/7501 |
<1/99802000 1/96064 1/2280304 |
| RMRP | AR | Transtornos do Espectro Hipoplasia Cartilagem-Cabelo com Displasia Anauxética (+) | Geral Amish Finlandês |
<1/500 <1/500 <1/500 |
99% 99% 99% |
<1/49901 <1/49901 <1/49901 |
<1/99802000 <1/99802000 <1/99802000 |
| RPE65 | AR | Amaurose Congénita de Leber 2 (+) | Geral | 1/228 | 98% | 1/11351 | 1/10352112 |
| RPE65 | AR | Retinite Pigmentosa 20 (+) | Geral | 1/228 | 98% | 1/11351 | 1/10352112 |
| RPE65 | AR | Doenças Relacionadas com RPE65 | Geral | 1/228 | 98% | 1/11351 | 1/10352112 |
| RS1 | XL | Retinosquise Juvenil: Ligada ao X | Geral | 1/2500 | 96% | 1/62476 | 1/249904 |
| RTEL1 | AR | Disqueratose Congénita Tipo 5 | Geral Judeu Ashkenazi |
1/500 1/203 |
99% 99% |
1/49901 1/20201 |
1/99802000 1/16403212 |
| SACS | AR | Ataxia Espástica Autossómica Recessiva de Charlevoix-Saguenay | Geral Franco-Canadense |
<1/500 1/19 |
95% 95% |
<1/9981 1/361 |
<1/19962000 1/27436 |
| SEPSECS | AR | Hipoplasia Ponto-Cerebelar Tipo 2D | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| SERPINA1 | AR | Deficiência de Alfa-1-Antitripsina | Geral Europeu |
1/33 1/19 |
95% 95% |
1/641 1/361 |
1/84612 1/27436 |
| SGCA | AR | Distrofia Muscular de Cinturas: Tipo 2D | Geral Europeu Finlandês |
<1/500 1/288 1/150 |
98% 98% 98% |
<1/24951 1/14351 1/7451 |
<1/49902000 1/16532352 1/4470600 |
| SGCB | AR | Distrofia Muscular de Cinturas: Tipo 2E | Geral Europeu |
1/500 1/406 |
98% 98% |
1/24951 1/20251 |
1/49902000 1/32887624 |
| SGCD | AR | Distrofia Muscular de Cinturas: Tipo 2F | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| SGCG | AR | Distrofia Muscular de Cinturas: Tipo 2C | Geral Marroquino Cigano |
1/381 1/250 1/96 |
98% 98% 98% |
1/19001 1/12451 1/4751 |
1/28957524 1/12451000 1/1824384 |
| SGSH | AR | Mucopolissacaridose Tipo IIIA (Síndrome de Sanfilippo Tipo A) | Geral Europeu |
1/454 1/253 |
98% 98% |
1/22651 1/12601 |
1/41134216 1/12752212 |
| SLC12A3 | AR | Síndrome de Gitelman | Geral | 1/100 | 98% | 1/4951 | 1/1980400 |
| SLC12A6 | AR | Síndrome de Andermann | Geral Franco-Canadense |
<1/500 1/23 |
98% 99% |
<1/24951 1/2201 |
<1/49902000 1/202492 |
| SLC17A5 | AR | Transtorno do Armazenamento do Ácido Siálico | Geral Finlandês |
<1/500 1/100 |
91% 91% |
<1/5545 1/1101 |
<1/11090000 1/440400 |
| SLC22A5 | AR | Deficiência Primária Sistémica de Carnitina | Geral Africano Afro-americano Este da Ásia Feroês Ilhas do Pacífico Sul da Ásia / Índia |
1/129 1/86 1/86 1/77 1/9 1/37 1/51 |
76% 76% 76% 76% 76% 76% 76% |
1/534 1/355 1/355 1/318 1/34 1/151 1/209 |
1/275544 1/122120 1/122120 1/97944 1/1224 1/22348 1/42636 |
| SLC25A13 | AR | Deficiência de Citrina | Geral Este da Ásia |
<1/500 1/65 |
95% 95% |
<1/9981 1/1281 |
<1/19962000 1/333060 |
| SLC25A15 | AR | Síndrome de Hiperornitinemia-Hiperamonémia-Homocitrulinúria (Síndrome do Triplo H) | Geral Franco-Canadense |
<1/500 1/37 |
99% 99% |
<1/49901 1/3601 |
<1/99802000 1/532948 |
| SLC25A20 | AR | Deficiência de Carnitina-Acilcarnitina Translocase | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| SLC26A2 | AR | Acondrogénese Tipo IB (+) | Geral Finlandês |
1/158 1/50 |
90% 90% |
1/1571 1/491 |
1/992872 1/98200 |
| SLC26A2 | AR | Atelosteogénese II (+) | Geral Finlandês |
1/158 1/50 |
90% 90% |
1/1571 1/491 |
1/992872 1/98200 |
| SLC26A2 | AR | Displasia Diastrófica (+) | Geral Finlandês |
1/158 1/50 |
90% 90% |
1/1571 1/491 |
1/992872 1/98200 |
| SLC26A2 | AR | Displasia Epifisária Múltipla 4 (+) | Geral Finlandês |
1/158 1/50 |
90% 90% |
1/1571 1/491 |
1/992872 1/98200 |
| SLC26A2 | AR | Doenças Relacionadas com SLC26A2 | Geral Finlandês |
1/158 1/50 |
90% 90% |
1/1571 1/491 |
1/992872 1/98200 |
| SLC26A3 | AR | Cloridrorreia Congénita Familiar | Geral Médio Oriente |
<1/500 1/57 |
98% 98% |
<1/24951 1/2801 |
<1/49902000 1/638628 |
| SLC26A4 | AR | Síndrome de Pendred | Geral Africano Afro-americano Europeu Este da Ásia |
1/80 1/76 1/76 1/88 1/74 |
98% 98% 98% 98% 98% |
1/3951 1/3751 1/3751 1/4351 1/3651 |
1/1264320 1/1140304 1/1140304 1/1531552 1/1080696 |
| SLC35A3 | AR | Artrogripose, Atraso Mental e Convulsões | Geral Judeu Ashkenazi |
<1/500 1/453 |
98% 98% |
<1/24951 1/22601 |
<1/49902000 1/40953012 |
| SLC37A4 | AR | Doença por Armazenamento de Glicogénio Tipo 1B | Geral Judeu Ashkenazi |
1/158 1/71 |
95% 95% |
1/3141 1/1401 |
1/1985112 1/397884 |
| SLC39A4 | AR | Acrodermatite Enteropática | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| SLC3A1 | AR, DG | Cistinúria Tipo I | Geral Europeu |
1/50 1/42 |
98% 98% |
1/2451 1/2051 |
1/490200 1/344568 |
| SLC45A2 | AR | Albinismo Oculocutâneo Tipo IV | Geral Japonês |
1/159 1/146 |
98% 98% |
1/7901 1/7251 |
1/5025036 1/4234584 |
| SLC4A11 | AR | Distrofia Endotelial da Córnea | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| SLC7A7 | AR | Intolerância Lisinúrica | Geral Finlandês Japonês |
<1/500 1/122 1/119 |
95% 95% 95% |
<1/9981 1/2421 1/2361 |
<1/19962000 1/1181448 1/1123836 |
| SLC7A9 | AR, DG | Cistinúria não Tipo I | Geral | 1/42 | 98% | 1/2051 | 1/344568 |
| SMN1 | AR | Atrofia Muscular Espinal | Geral Africano Afro-americano Judeu Ashkenazi Europeu Este da Ásia Latino |
1/54 1/72 1/72 1/67 1/47 1/59 1/68 |
91% 71% 71% 91% 95% 93% 90% |
1/590 1/246 1/246 1/734 1/921 1/830 1/671 |
1/127440 1/70848 1/70848 1/196712 1/173148 1/195880 1/182512 |
| SMPD1 | AR | Doença de Niemann-Pick: Tipo A/B | Geral Judeu Ashkenazi Latino |
1/250 1/115 1/106 |
95% 95% 95% |
1/4981 1/2281 1/2101 |
1/4981000 1/1049260 1/890824 |
| SRD5A2 | AR | Deficiência de 5-Alfa Redutase | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| STAR | AR | Hiperplasia Suprarrenal Congénita Lipóide | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| SUMF1 | AR | Deficiência Múltipla de Sulfatase | Geral Judeu Ashkenazi |
1/500 1/320 |
98% 98% |
1/24951 1/15951 |
1/49902000 1/20417280 |
| TAT | AR | Tirosinemia, Tipo II | Geral | 1/250 | 98% | 1/12451 | 1/12451000 |
| TCIRG1 | AR | Osteoporose Relacionada com TCIRG1 | Geral | 1/250 | 98% | 1/12451 | 1/12451000 |
| TECPR2 | AR | Paraparésia Espástica 49 | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| TFR2 | AR | Hemocromatose Tipo III | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| TGM1 | AR | Ictiose Congénita | Geral | 1/224 | 95% | 1/4461 | 1/3997056 |
| TH | AR | Síndrome de Segawa | Geral | 1/224 | 98% | 1/11151 | 1/9991296 |
| TMEM216 | AR | Síndrome de Joubert 2 (+) | Geral Judeu Ashkenazi |
1/141 1/92 |
98% 98% |
1/7001 1/4551 |
1/3948564 1/1674768 |
| TMEM216 | AR | Síndrome de Meckel 2 (+) | Geral Judeu Ashkenazi |
1/141 1/92 |
98% 98% |
1/7001 1/4551 |
1/3948564 1/1674768 |
| TMEM216 | AR | Doenças Relacionadas com TMEM216 | Geral Judeu Ashkenazi |
1/141 1/92 |
98% 98% |
1/7001 1/4551 |
1/3948564 1/1674768 |
| TPP1 | AR | Lipofuscinose Ceróide Neuronal, Relacionada com TPP1 | Geral Franco-Canadense |
1/252 1/53 |
97% 97% |
1/8368 1/1734 |
1/8434944 1/367608 |
| TRIM32 | AR | Síndrome de Bardet-Biedl 11 (+) | Geral Huterita |
<1/500 1/12 |
98% 98% |
<1/24951 1/551 |
<1/49902000 1/26448 |
| TRIM32 | AR | Distrofia Muscular de Cinturas Tipo 2H (+) | Geral Huterita |
<1/500 1/12 |
98% 98% |
<1/24951 1/551 |
<1/49902000 1/26448 |
| TRIM32 | AR | Doenças Relacionadas com TRIM32 | Geral Huterita |
<1/500 1/12 |
98% 98% |
<1/24951 1/551 |
<1/49902000 1/26448 |
| TRMU | AR | Insuficiência Hepática Aguda, Infantil | Geral Judeu Iemenita |
<1/500 1/34 |
98% 98% |
<1/24951 1/1651 |
<1/49902000 1/224536 |
| TSEN54 | AR | Hipoplasia Ponto-Cerebelar Relacionada com TSEN54 | Geral | 1/250 | 98% | 1/12451 | 1/12451000 |
| TTC37 | AR | Síndrome Trico-Hepato-Entérica | Geral | 1/500 | 98% | 1/24951 | 1/49902000 |
| TTPA | AR | Ataxia com Deficiência Isolada de Vitamina E | Geral Europeu |
<1/500 1/267 |
98% 90% |
<1/24951 1/2661 |
<1/49902000 1/2841948 |
| TYMP | AR | Encefalopatia Neurogastrointestinal Mitocondrial | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| TYR | AR | Albinismo Oculocutâneo Tipo I | Geral | 1/100 | 98% | 1/4951 | 1/1980400 |
| TYRP1 | AR | Albinismo Oculocutâneo Tipo III | Geral Africano |
<1/500 1/47 |
98% 98% |
<1/24951 1/2301 |
<1/49902000 1/432588 |
| UGT1A1 | AR | Síndrome de Crigler-Najjar | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| USH1C | AR | Surdez não Sindrómica Relacionada com USH1C (+) | Geral Franco-Canadense |
1/353 1/227 |
90% 90% |
1/3521 1/2261 |
1/4971652 1/2052988 |
| USH1C | AR | Síndrome de Usher Tipo 1C (+) | Geral Franco-Canadense |
1/353 1/227 |
90% 90% |
1/3521 1/2261 |
1/4971652 1/2052988 |
| USH1C | AR | Doenças Relacionadas com USH1C | Geral Franco-Canadense |
1/353 1/227 |
90% 90% |
1/3521 1/2261 |
1/4971652 1/2052988 |
| ISH2A | AR | Síndrome de Usher Tipo 2A | Geral Europeu |
1/126 1/73 |
96% 96% |
1/3126 1/1801 |
1/1575504 1/525892 |
| VPS13A | AR | Coreoacantocitose | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| VPS13B | AR | Síndrome de Cohen | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| VPS53 | AR | Hipoplasia Ponto-Cerebelar: Relacionada com VPS53 | Geral Judeu Marroquino |
<1/500 1/37 |
98% 98% |
<1/24951 1/1801 |
<1/49902000 1/266548 |
| VRK1 | AR | Hipoplasia Ponto-Cerebelar Tipo 1A: Relacionada com VRK1 | Geral | <1/500 | 98% | <1/24951 | <1/49902000 |
| VSX2 | AR | Microftalmia com ou sem Coloboma | Geral | 1/91 | 98% | 1/4501 | 1/1638364 |
| WAS | XL | Neutropénia Congénita Severa Relacionada com WAS (+) | Geral | 1/125000 | 99% | 1/12499901 | 1/49999604 |
| WAS | XL | Trombocitopenia: Ligado ao X (+) | Geral | 1/125000 | 99% | 1/12499901 | 1/49999604 |
| WAS | XL | Síndrome de Wiskott-Aldrich (+) | Geral | 1/125000 | 99% | 1/12499901 | 1/49999604 |
| WAS | XL | Doenças Relacionadas com WAS | Geral | 1/125000 | 99% | 1/12499901 | 1/49999604 |
| WRN | AR | Síndrome de Werner | Geral Europeu Japonês |
1/308 1/112 1/71 |
98% 98% 98% |
1/15351 1/5551 1/3501 |
1/18912432 1/2486848 1/994284 |
| XPA | AR | Xeroderma Pigmentosa: Grupo A | Geral Japonês |
1/500 1/74 |
99% 99% |
1/49901 1/7301 |
1/99802000 1/2161096 |
| XPC | AR | Xeroderma Pigmentosa: Grupo C | Geral | 1/500 | 99% | 1/49901 | 1/9980200 |
* Em genes com resultado negativo.
** Em genes com resultado negativo e parceiro não analisado.
Abreviaturas: AR, Autossómica Recessiva; XL, Ligado ao X; DG, Digénico
O princípio genético:
As células são os “tijolos” que formam o nosso organismo. Cada uma de nossas células contém toda a nossa informação genética, sob a forma de genes que, por sua vez, se encontram empacotados em cromossomas. Os genes dão as instruções que permitem ao nosso corpo crescer e funcionar. O ser humano tem 46 cromossomas que estão agrupados aos pares. Os primeiros 22 pares são numerados de 1 a 22. Ao último par, chamamos cromossoma X e cromossoma Y (cromossomas sexuais) e é este último par que define o género feminino ou masculino de um indivíduo.
Herdamos sempre um cromossoma da mãe e um cromossoma do pai do mesmo modo que genes também são herdados aos pares. Herdamos uma cópia de cada par de genes da nossa mãe e a outra cópia do nosso pai. Os genes são formados por DNA, e o DNA é formado por duas cadeias que se enrolam como uma escada torcida. As cadeias de DNA são compostas por quatro letras que se repetem numa determinada sequência ou ordem. Essas quatro letras A, T, G e C são chamadas de bases.
Algumas doenças ocorrem quando há uma variante (mutação) no DNA. Uma mutação acontece quando as bases que compõem o DNA são alteradas ou "mutadas" em relação à sequencia de referência do genoma humano. Uma mutação patogénica faz com que o gene funcione incorretamente ou simplesmente não funcione. As doenças genéticas acontecem quando um ou mais genes não funcionam corretamente.
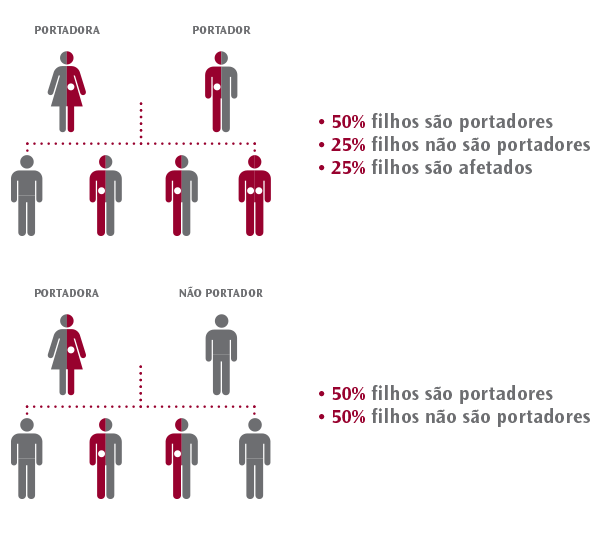
Hereditariedade autossómica recessiva:
Existem diferentes maneiras pelas quais as doenças genéticas podem ser transmitidas ou herdadas nas famílias. Algumas doenças genéticas aparecem quando apenas a cópia materna ou a cópia paterna do gene tem uma mutação. São as chamadas doenças genéticas dominantes. O teste de portadores não está indicado para determinar o risco de transmissão de doenças genéticas dominantes. Outro tipo de doenças genéticas ocorre quando ambas as cópias de um par de genes têm uma mutação, a cópia materna e a cópia paterna. Essas doenças são chamadas de doenças genéticas recessivas. Um portador de uma doença genética recessiva é alguém que tem uma mutação num dos genes associado a uma doença recessiva.
Um casal pode ter um filho com doença recessiva quando a mãe e o pai são portadores de uma mutação no mesmo gene que causa a doença. Em cada gravidez, a probabilidade desse casal ter um filho com a doença é de 1 em 4, ou seja, é de 25%. Esse casal também tem uma probabilidade de 3 em 4, ou seja, de 75%, de ter um filho que NÃO tenha a doença.
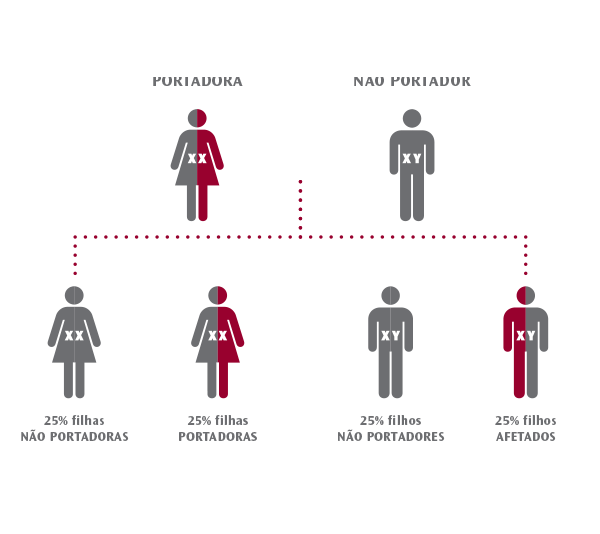
Hereditariedade ligada ao cromossoma X:
O sexo masculino ou feminino é definido pelos “cromossomas sexuais” X e Y. As mulheres têm duas cópias do cromossoma X. Os homens têm um cromossoma X e um cromossoma Y. Isso significa que as mulheres têm duas cópias de cada um dos genes do cromossomo X e os homens têm apenas uma cópia desses genes. Algumas doenças genéticas são causadas por mutações em genes que se encontram no cromossoma X. Essas doenças são chamadas de doenças genéticas ligadas ao X. As mulheres podem ser portadoras de doenças ligadas ao X. Essas mulheres têm uma cópia funcional e uma cópia não funcional de um gene ligado ao X. Os homens não são, em regra, portadores porque têm apenas um cromossoma X, portanto, serão saudáveis ou afetados (doentes).
Para que as doenças genéticas ligadas ao X se manifestem, a mãe precisa de ser portadora para haver probabilidade de ter um filho afetado. A probabilidade dessa mulher, em cada gravidez, ter um filho afetado pela doença é de 25% (ou 1 em 4). A mesma mulher tem 25% de probabilidade, em cada gravidez, de ter uma filha portadora. Do mesmo modo, a mesma mulher, tem 75% de probabilidade de ter um filho que NÃO tenha a doença.

O que posso fazer se for portadora de uma mutação num gene que cause doença genética recessiva?
Quando planear uma gravidez, o seu parceiro deverá ser testado para perceber se ele próprio é também portador de uma mutação no mesmo gene. Se o seu parceiro também for portador de uma mutação no mesmo gene, existem diferentes opções reprodutivas a ser consideradas.
Para muitos casais, saber o seu estado de portador antes de serem pais permite que tomem decisões reprodutivas informadas que podem ter impacto no futuro dos seus filhos. Casais em risco de ter um bebé com uma doença recessiva ou ligada ao X têm a oportunidade de considerar:
1
Uma gravidez natural e optar por fazer diagnóstico pré-natal através do estudo genético de vilosidades coriónicas ou do liquido amniótico.
2
Realizar um teste genético pré-implantação (PGT) com fertilização in vitro (FIV) para poder selecionar os embriões geneticamente não afetados e, em seguida, transferir embriões livres da doença.
3
Recorrer à utilização de espermatozoides ou óvulos de dadores.
Qualquer dúvida contacte o Centro de Medicina Laboratorial
Germano de Sousa e agende uma consulta de Genética Médica
CONSULTA DE
GENÉTICA MÉDICA
ACONSELHAMENTO PRÉ E PÓS TESTE
Disponível em Lisboa e Porto
